Since its first formal description in 1958, the medical understanding of and clinical approach toward hypertrophic cardiomyopathy (HCM) has evolved significantly.
Not surprisingly, the terminology used to describe the condition has evolved as well. Once thought to be a rare, highly lethal and untreatable disease with an annual mortality rate as high as 6% per year, it is now known to be a common familial condition with only modest mortality (approximately 1%) in most cases.
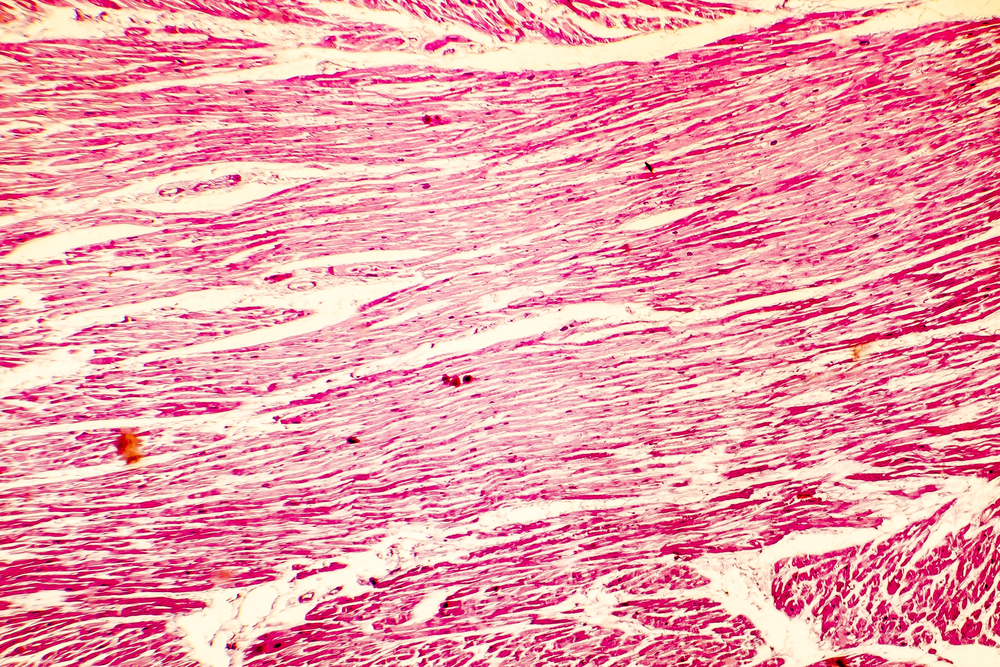
In the January 1958 edition of the British Heart Journal, pathologist Donald Teare reported eight cases of sudden cardiac death in adolescents or young adults. Upon microscopic examination of the hearts at autopsy, all were found to have striking septal hypertrophy and a “bizarre arrangement of bundles of muscle fibres running in diverse directions and separated by connective tissue and clefts.”1 Teare postulated that the cause was a type of diffuse tumor that he called a “muscular hamartoma.”
Extensive research over the last 50 years, helped considerably by the development of echocardiography in the early 1970s, has provided a much broader and deeper understanding of this condition while validating many of Teare’s original observations. Rather than a tumor, this condition is now recognized as a genetically mediated primary disease of the myocardium – that is, a cardiomyopathy. Initially referred to by terms such as idiopathic hypertrophic subaortic stenosis (IHSS), hypertrophic obstructive cardiomyopathy (HOCM), muscular subaortic stenosis, and asymmetric septal hypertrophy (ASH), it has since been recognized that the condition can present in a variety of ways: not all cases affect the septum disproportionately, and about one-third of cases do not have obstruction to left ventricular outflow. Therefore the more general term “hypertrophic cardiomyopathy” (or HCM) is now preferred.
Definition, Epidemiology, and Cause
HCM is a disease found worldwide with a fairly consistent global prevalence of 0.2% (or 1 in 500). It is caused by pathogenic variations in at least one of the eight genes that encode for protein constituents of the sarcomere, which is the contractile unit of cardiac muscle. The disease is inherited in an autosomal dominant pattern; however, there is significant variation in expression even within families with the same mutation. At this point, more than 1,400 HCM-causing genetic mutations have been identified and novel mutations are common6.
The current generally accepted definition of HCM is a hypertrophied and nondilated left ventricle in the absence of other cardiac conditions (such as uncontrolled hypertension and aortic valve disease) that could also produce the findings. The hypertrophy may be quite focal or more generalized, and usually (but not always) has a wall thickness of ≥15 mm in adults (13-14 mm is considered borderline). In children, wall thickness of ≥2 standard deviations above the mean for age, sex, or body size meets the definition. MRI studies have shown that the left ventricular anterior free wall and contiguous basal anterior septum are the most frequently involved segments. A significant number of cases have involvement limited to areas of the left ventricle not well seen by echocardiography4.
The increased availability of genetic testing is identifying a growing population of individuals known to carry a pathogenic mutation (genotype-positive) but without clear clinical manifestations of the disease. These manifestations are referred to either as phenotype-negative or hypertrophy-negative. Research is also indicating that there are certain structural changes aside from hypertrophy that may indicate a “subclinical” or “preclinical” state.
Clinical Manifestation
HCM is often asymptomatic, especially when there is no obstruction to left ventricular outflow. When present, symptoms may include palpitations, dyspnea, fatigue, and syncope, particularly following exertion. Sudden cardiac death due to ventricular arrhythmia may be the initial manifestation of the disease. Atrial fibrillation, heart failure, and thromboembolic events are also potential complications. The ECG is usually abnormal with findings suggestive of left ventricular hypertrophy, such as increased QRS voltage and ST-T wave changes, but it may also be completely normal in a small minority of cases.
Echocardiography is the mainstay for diagnosis and risk stratification, although there is an increasing role for MRI. In addition to a thickened, nondilated ventricle, the ejection fraction is often high despite evidence of diastolic dysfunction. There may be systolic anterior motion (SAM) of the mitral valve with eccentric mitral regurgitation and a dynamic left ventricular outflow tract gradient.
An MRI may enable a more accurate assessment of wall thickness in all areas of the ventricles and can also assess for fibrosis, which is an emerging marker for increased arrhythmia risk.
Clinical expression of HCM usually increases with age. Hypertrophy most often becomes apparent during adolescence, but a first appearance later in life is not uncommon. ECG changes, increased ejection fraction, and delayed myocardial relaxation may precede the onset of hypertrophy.
Mortality and Risk Stratification
The natural history of HCM is varied and unpredictable. Early studies from tertiary care centers reported adult HCM mortality as high as 6% per year. However, more recent research from community-based HCM populations has shown much more favorable outcomes, with mortality of less than 1% per year. The difference is thought to be due primarily to selection bias, with the tertiary care cohorts being more severely affected than the community-based cohorts. Mortality in children with HCM may be 2% per year or higher.
Despite this overall more optimistic view of HCM outcomes, there is a subset of those with HCM who are at significantly higher risk for complications such as sudden cardiac death and progression to heart failure. Although a number of risk markers have been identified, no universally accepted risk prediction model yet exists, and identifying those at increased risk remains imprecise.
The 2011 American College of Cardiology Foundation/American Heart Association Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy lists the following items as established or potential markers of increased arrhythmia risk6:
Other than previous cardiac arrest, established risk markers have fairly low positive predictive values (between 10% and 20%). Most HCM centers in the U.S., however, will consider placement of an implanted cardioverter/defibrillator (ICD) when one or two risk markers are present.
 Because of their concern that the U.S. risk stratification approach overestimates risk, resulting in inappropriate ICD placements, a European group (the Hypertrophic Cardiomyopathy Outcomes Investigators) developed a risk prediction model that allows for weighting of factors and use of continuous (rather than binary) variables. The HCM Risk-SCD model is intended as a primary prevention risk stratifier – those with a personal history of cardiac arrest are excluded.
Because of their concern that the U.S. risk stratification approach overestimates risk, resulting in inappropriate ICD placements, a European group (the Hypertrophic Cardiomyopathy Outcomes Investigators) developed a risk prediction model that allows for weighting of factors and use of continuous (rather than binary) variables. The HCM Risk-SCD model is intended as a primary prevention risk stratifier – those with a personal history of cardiac arrest are excluded.
The model, which provides an estimate of the absolute risk of sudden death within the next five years, utilizes the following factors7:
- Age
- Maximal left ventricular wall thickness
- Left atrial diameter
- Left ventricular outflow gradient
- Family history of HCM and sudden death
- Nonsustained ventricular tachycardia
- Unexplained syncope
This approach, however, has been criticized for being unreliable for the classification of both high- and low-risk cases8. At this time, risk stratification requires consideration of multiple factors and remains an imprecise process.
Management
All cases of HCM should be assessed for arrhythmia risk, even those who are completely asymptomatic. Individuals should avoid strenuous physical exercise and other cardiovascular risk factors and comorbidities need to be treated aggressively. In addition, it is recommended that family members be screened by having echocardiograms and ECGs usually beginning around age 12 and continuing periodically throughout life.
Therapeutic options have become widely available over the last two decades for those at increased arrhythmia risk and for those with symptomatic obstruction. ICDs have been shown to successfully terminate ventricular arrhythmias; however there is a significant complication rate and long-term mortality data is not yet available. Likewise, septal reduction procedures such as myectomy and alcohol septal ablation can be effective in reducing obstructive symptoms but long-term mortality data is sparse.
Underwriting Challenges
Although there have been many advances in the field of HCM, many challenges remain both for the clinician and the insurance underwriter. Some important issues for an insurance underwriter to consider include the following:
- Since HCM is often asymptomatic, insurance applicants may be unaware of the condition, yet abnormalities may be recognized in medical records or in tests obtained in conjunction with the application.
- Mild or borderline hypertrophy may be attributed to other causes without full clinical investigation.
- Maximum wall thickness may occur in areas not well seen by echocardiography.
- A family history of HCM may be disclosed by a proposed insured; however, clinical evaluation may not have been completed.
- With more genetic testing and family screening, the population of those known to be genotype-positive hypertrophy-negative will increase, and the long-term outcome for these “preclinical” states requires more study.
Conclusion
The broad clinical spectrum of HCM was well-represented in Teare’s original group of eight cases. With intense research and enhanced diagnostic tools, particularly echocardiography and more recently magnetic resonance imaging, much has been learned about it. However, there are still many uncertainties. Also, because of HCM’s recognized high prevalence and potential for significantly increased mortality, insurance underwriters must be familiar with contemporary risk stratification approaches. The recommendation for family screening and the availability of genetic testing has added layers of complexity to this already difficult underwriting challenge.
References