Myocardial infarction is a major trigger for living benefits claims and the survivability of infarction is such that a history is increasingly common in applicants.
Medical definitions of infarction have changed markedly over the last century, from what was initially a postmortem diagnosis of a condition that was considered inevitably fatal to a condition defined by circulating biological markers and where survival is the expectation.
Population demographics and the evolution of concepts as to how infarction should be defined and classified in various clinical settings have resulted in changes in incidence that have driven a need to continually monitor and revise pricing, underwriting, and claims models.
The basic tenet that a diagnosis of infarction is made when death of myocardial cells occurs as a result of inadequate blood supply has not changed, but assays of biological markers that reflect cell death have become surrogates for histopathological proofs.
Central to the triage of patients with coronary syndromes is a capacity to reliably distinguish unstable angina from infarction. The improved analytical discriminatory power of assays of circulating biological markers over time has resulted in a diagnosis of infarction being assigned to some who would previously have been diagnosed otherwise. The capacity to detect smaller and smaller infarcts than was previously possible has changed the relative proportions of those labeled infarction and unstable angina.
This article traces the history of the definition of infarction, documents the current standardized position, and looks at the potentials for change as technologies advance and medical opinion evolves.
Until the end of the 19th century infarction was considered a fatal entity only diagnosable post-mortem. The diagnosis required evidence of thrombotic occlusion of a coronary artery in the context of sudden death, or death within a short period of time following the onset of chest pain.
That prevailing concept was re-examined by Dr. James Herrick in 1912 in the first classification of the clinical features of coronary obstruction. Herrick disputed the long-held view that infarction was inevitably fatal in providing clinical evidence that it was often survived without necessarily being associated with major morbidity.1, 2 Acceptance of that view drove a need to define reliable parameters for diagnosing infarction in those with chest pain who had not succumbed to sudden death and who presented to medical attention.
In Herrick’s time the diagnosis was based purely on clinical grounds with ischemic chest pain being the usual criterion. ECG accompaniments became supporting criteria following their description by Herrick in 1919.15 In that era, mortality rates were of the order of 30% and treatment options were limited to weeks of bed rest. There was no material pharmacological capacity to influence prognosis and the resumption of a normal lifestyle with return to work was discouraged.
By the 1950s and 1960s myocardial infarction had become the most common cause of death in Europe and North America. The health and socioeconomic burden resulted in the establishment of dedicated coronary care units and work on pharmacological and interventional strategies that had the capacity to reduce mortality and limit infarct size. That focus saw the creation of protocols that enabled earlier discharge and resulted in reductions in mortality from 30% to 15% over the next few decades.3
Strategies which improved outcomes mandated reliable parameters for diagnosis. It was recognized that standardized and universally accepted definitions would provide benchmarks by which emerging treatments could be compared and judged with statistical certainty. Recognizing the need for such protocols in the clinical and research setting, the World Health Organization (WHO) published the first guideline for the definition of myocardial infarction in 1959. That definition was based on ischemic pain and ECG changes.
In 1979, WHO published a revision that allowed the detection of elevated cardiac enzymes in the circulation to be included as a third criterion although elevated enzymes were not mandated for a diagnosis. Given the limited sensitivity of the ECG in the more common non-ST elevation ECG syndromes, this revision resulted in an increase in incidence. A diagnosis was now possible in those with ischemic pain who did not satisfy ECG requirements.
Despite the benefits of including enzymes as markers of cell death, definitions remained very imperfect by today’s standards. Enzymes lacked good sensitivity for damage and there was a lack of specificity because these enzymes were not absolutely cardiac-restricted. Coupled with the fact that there was no analytic standardization across various enzyme assays, levels were required to rise to twice upper reference limits (URL) to reduce false positives.
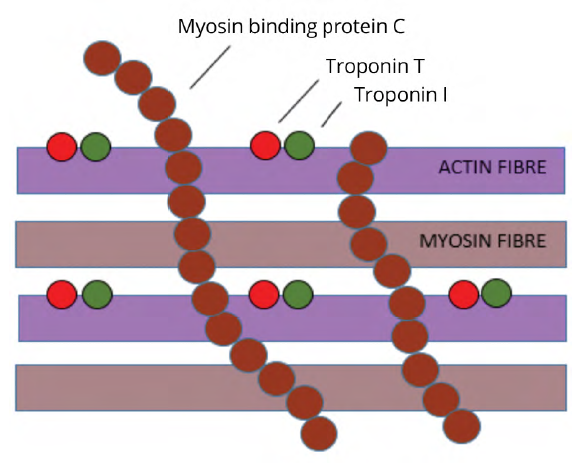
During the 1970s and 1980s there was research interest in the structure of actin and myosin, the two filaments in heart muscle cells responsible for cell contraction. Two proteins associated with the thin actin filament (contractile proteins troponin I and troponin T) were identified and were shown to be cardiac-specific. These proteins were undetectable in the blood of normal populations at the time and there was interest in determining if they were released into the circulation in detectable amounts following cell damage.
A protein associated with the thick myosin filament (myosin binding protein c) was also identified and also shown to be cardiac-specific. Research interest in that protein became directed towards the identification of the mutations in that protein that had become implicated in the development of cardiomyopathy. As such that protein was not pursued as a potential marker for ischemic damage at that time (Fig 1).4
Figure 1:
Components of Human Cardiac Muscle
Troponin I and troponin T were able to be detected in those infarcts defined using enzyme criteria with very reliable accuracy and with some improvements in sensitivity. The high level of sensitivity coupled with the specificity benefits provided momentum for further research.
By the mid-1990s a subset of acute coronary syndromes not assigned a diagnosis of infarction on enzyme criteria but which had detectable troponins was defined. That subset was demonstrably at high risk on the basis of 30-day mortality. (Fig 2).5
Figure 2:
30-Day Mortality in Acute Coronary Syndromes5

Universal Definition of Myocardial Infarction
Strong clinical imperatives promoted discussions that troponins be considered the preferable markers for risk given that enzyme-based determinates of cell death did not provide acceptable prognostic triage. It was estimated that a transition from enzymes to troponins as the benchmark marker might increase the prevalence of infarction in coronary syndromes from 30% to as high as 80%.
The transition from enzymes to troponins as preferred markers was ratified in a Universal Definition of Myocardial Infarction that was published in 2000. That guideline also contained a paradigm shift in thinking with a recommendation that biomarker evidence become fundamental and central to the diagnosis. A cut point for an elevated troponin level was assigned arbitrarily at the 99th percentile for a reference population. That cut point was approximately three standard deviations (SDs) from the mean and was chosen to minimize false positives.
Guidelines recommended not only that one troponin level exceed the 99th percentile but that a typical release pattern be confirmed by the detection of rising and/or falling levels. Evidence of appropriate troponin dynamics was mandated to help confirm the clinical context and to avoid false positives in those with chronically elevated biomarkers and non-ischemic conditions. Implementation of those guidelines provided better detection, more prompt triage to treatment and a further fall in mortality to around 3%.
Mortality rates were also influenced by the improving analytic sensitivity of markers which increased the detection of small and less immediately life threatening infarcts and with the inclusion of these cases in outcome statistics.
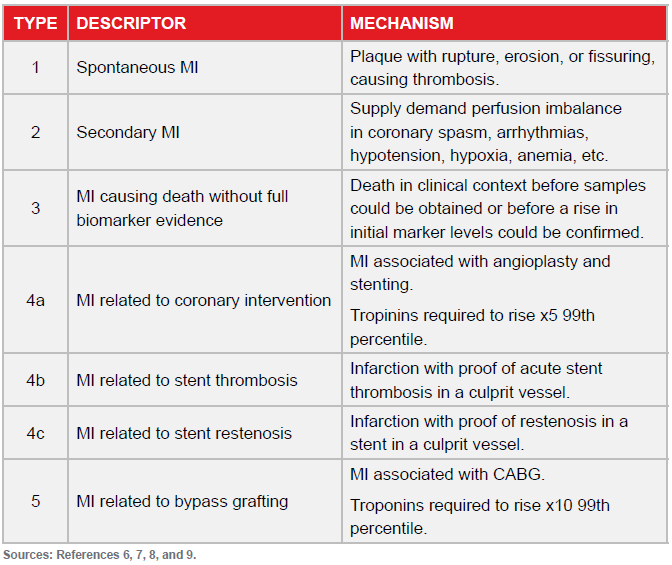
The Universal Definition was modified in 2007 and again in a third iteration in 2012.6, 7 Based on etiology, five categories of infarction were described and which allowed a diagnosis outside of the traditional plaque rupture with thrombosis setting (Fig 3).
Figure 3:
Types of Myocardial Infarction
Since troponins were endorsed as preferred biomarkers there have been five generations of assays, and each analytic refinement has provided improvements in sensitivity for diagnosis and a capacity to detect troponins in normal populations. The emerging high sensitivity assays are required to be capable of detecting troponins in at least 50% of normal people with very high degrees of precision at the 99th percentile.
While early assay results were highly specific they were relatively insensitive. Much emphasis needs to be placed on the fact that increases in sensitivity occur at the expense of specificity. While troponins are markers of cell death they do not indicate the cause of damage and the specificity concerns for high sensitivity assays loom large.
With a diagnosis of infarction only possible when biomarkers rise as a consequence of ischemia it is imperative that clinical histories be clarified and that evidence supporting the requirement that cell death resulted from lack of blood supply be provided.
In the majority of cases where elevated levels of troponins are detected in emergency departments the discharge diagnosis is not infarction but another condition that has the capacity to damage myocytes. The causes of non-ischemic elevations of troponins can be found elsewhere.8
The Third Universal Definition 2012 included the following implications and recommendations:9
- Emphasis was placed on biomarkers as the cornerstone for diagnosis with appropriate biomarker dynamics being satisfied on serial sampling.
- The impact of troponins increasing incidence of myocardial infarction was confirmed as being between 41% (Minnesota, U.S.) and 83% (Finland).
- Advocacy for the 99th percentile for the URL was confirmed with that decision limit having the capacity to maximize detection while minimizing false positives.
- A concept of injury was introduced to emphasize that cell death was not synonymous with infarction. It was stressed that small amounts of cell necrosis may be associated with conditions including heart failure, renal failure, myocarditis, sepsis and pulmonary embolism and as a result of otherwise uneventful percutaneous procedures or coronary surgical procedures. There was a recommendation that these should be labeled non-ischemic injury and not infarction.
- The previously defined five categories of infarction were ratified. There was an endorsement that infarction be diagnosed regardless of the mechanism by which ischemia was promoted and that that infarction may occur in people with normal coronary arteries. It was recognized that given the complexities of some situations it was not always easy to discriminate between non-ischemic injury and infarction.
Conclusions and Implications for Insurers
Definitions of myocardial infarction are in flux and open to change with some questions implicit in guidelines being unanswered or open to interpretation. The following points must all be considered when developing insurance products which provide myocardial infarction benefits:
- The degree to which troponins might have a sensitivity deficit for defining infarction remains uncertain and other potential markers continue to be tested. The most pressing indication for continued improvements in assay sensitivities and the search for new biological signals is to enable the exclusion of infarction earlier than is currently possible with serial troponin testing. Cardiac myosin binding protein has now been tested in that context. Its abundance in the cell results in its very early appearance in the circulation following necrosis and as such it holds promise in that regard. There is no evidence that it is more sensitive for diagnosis than troponin based protocols.14
- Despite mandating dynamic changes in markers for a diagnosis, definitions have not quantified what constitutes a significant change for that purpose. Clinical protocols require a relative change from baseline at presentation but there is a lack of consistency in the magnitude of change required for confirmation. Based on analytical variability a change of >20% has been considered adequate but some protocols mandate higher degrees of relative change.10
- Absolute changes in troponin levels may prove to have better diagnostic utility than relative changes. Diagnostic and triage approaches incorporating absolute change now appear in the literature and suggested magnitudes of change have been published.
- Absolute levels, whether above or below accepted 99th percentile thresholds, carry prognostic implications. With risk being continuous for all levels of troponin it can be argued that the 99th percentile lacks biological plausibility. A clinical risk focus based on absolute levels rather than arbitrary dichotomous labeling has
advantages. These notions leave open the potential for constructs of additional pathways incorporating lower biomarker thresholds for risk stratification and diagnosis in acute coronary syndromes where troponins do not reach the 99th percentile. - There is no standardization of what constitutes a normal population for the purposes of defining biomarker reference limits. Constituents of currently defined normal populations have diverse demographic characteristics and include both high and low cardiovascular risk individuals. Troponin levels are higher in males than in females across a range of assays. Gender-specific cutoffs may be endorsed and would increase incidence rates in women while providing a prognostic benefit to those women who might be currently underdiagnosed.11
- The focus on biomarkers is centered on the clinical need to rule in or rule out infarction early after presentation and not on a need to document infarct size. In clinical practice troponin levels are not commonly repeated after triage with infarct size tracking most accurately with levels taken at day three or four. Recommendations exist that suggest follow up levels might be assayed for that purpose.10
Author’s Note: Readers are referred to “High Sensitivity Troponin Assays: Answers to Frequently Asked Questions” for a more detailed exploration of some of these points and other frequently raised matters.13