Perhaps no other field of neurology has seen as rapid and promising developments as multiple sclerosis.
Projected improvements in mortality must be tempered by unique risks associated with new treatment modalities. Increasingly sensitive diagnostic tools, changing criteria, and trends toward treatment earlier in disease course will have a significant impact on critical illness assumptions. The purpose of this article is therefore to review changes in diagnostic criteria, emerging concepts in demyelinating variants, new imaging modalities, and a wave of new pharmacologic treatments.
Background
Multiple sclerosis (MS) is the prototypical autoimmune chronic demyelinating disorder. According to the recently released Atlas of Multiple Sclerosis,1 there are 2.3 million people living worldwide with the disease corresponding to a prevalence of 33 per 100,000. There are roughly 400,000 people in the United States with the disorder. The average age of onset is 30 years with a well-described and stable 2:1 female to male ratio. The previous 2008 report estimated global incidence at 2.5 per 100,000.2 An interesting and unique epidemiologic feature of MS is the decreasing prevalence rates closer to the equator, supporting emerging theories highlighting the role of Vitamin D (and sunlight) in the disorder. The ultimate etiology remains hotly debated. Most current pathophysiologic theories center around a combination of genetic susceptibility factors and environmental influences such as viral exposures. The end result is repeated demyelinating attacks on the axons of the brain and spinal cord, followed by remyelination and corresponding clinical improvement, almost certainly partial in nature. While multiple sclerosis is classically thought of as a demyelinating white mattDeer disorder, increasing emphasis is being placed upon axon loss in the disorder, grey-matter involvement, and subsequent prognostic implications.
Diagnostic Criteria
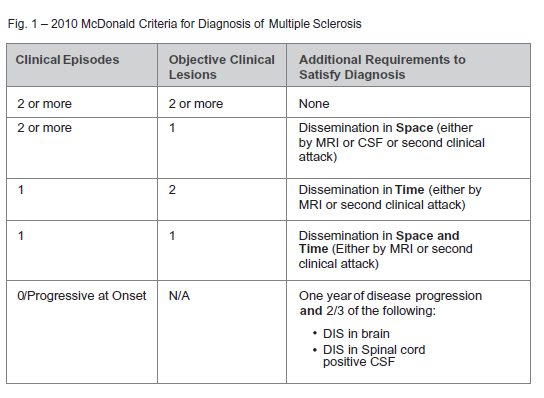
The initial challenge in multiple sclerosis remains accurate diagnosis as there is no single ‘gold standard’ diagnostic tool that offers complete certainty, especially early on in disease course. Accordingly, several criteria have been developed to solidify diagnosis. Originally developed in 2001, the McDonald criteria have garnered widest acceptance over time. The criteria have undergone major revisions in 2005 and in 2010. Of interest to the critical illness industry, with each revision, the criteria have become less ‘stringent’ in terms of objective radiographic and clinical requirements necessary to satisfy the diagnosis. At its core, the criteria remain centered about the oft-repeated “dissemination in time and space” (see Figure 1).
Dissemination In Space – One or more characteristic MS lesions in at least two of the following locations:
- Periventricular
- Juxtacortical
- Infratentorial
- Spinal Cord
Dissemination in Time
- A new characteristic enhancing and/or nonenhancing lesion on MRI in comparison with a baseline scan performed.
- Any combination of non- enhancing and asymptomatic enhancing MRI lesions on the same scan.
Key updates with Revised 2010 McDonald criteria.3
- Reduction in the number of MS lesions required to satisfy definition of Dissemination in Space. This can now be satisfied with a minimum of two lesions.
- Removal of 30-day time period previously required between first clinical attack and MRI of the brain to satisfy Dissemination in Time criteria.
- In some cases, Dissemination in Time may now be satisfied with a single scan
- Removal of CSF studies from DIS definition, further de-emphasizing their importance.
Developments in Imaging
Optical Coherence Technology
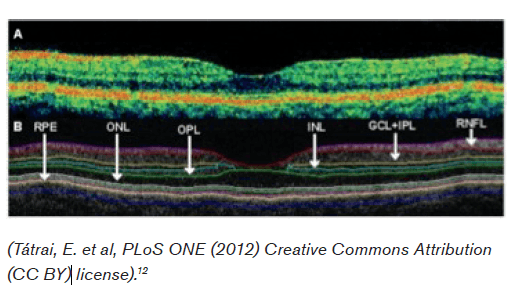 Optical Coherence Technology (OCT) is an increasingly utilized high-resolution form of computed tomography for the detection of retinal and optic nerve disorders. Infrared light is typically used to reconstruct the macular and peripapillary areas, producing an image reminiscent of B mode ultrasound.4 With a resolution of 5-7 micrometers it has been described as providing a near “in vivo ‘optical biopsy’” of the areas of interest.5 In equivocal, remote, or retrobulbar (i.e., not detectable by direct opthalmoscope) cases of optic neuritis, OCT can provide an objective and accurate measurement of retinal nerve fiber layer thickness. As discussed below, patients on Fingolimid (Gilenya) must be monitored for the development of macular edema, which can also be detected with this technology. Interestingly, axon loss in multiple sclerosis has been well demonstrated in the absence of clinical optic neuritis, thus prompting interest as a measure of overall disease activity and prognosis. OCT has not however been found to predict progression from isolated optic neuritis to clinically definite multiple sclerosis.
Optical Coherence Technology (OCT) is an increasingly utilized high-resolution form of computed tomography for the detection of retinal and optic nerve disorders. Infrared light is typically used to reconstruct the macular and peripapillary areas, producing an image reminiscent of B mode ultrasound.4 With a resolution of 5-7 micrometers it has been described as providing a near “in vivo ‘optical biopsy’” of the areas of interest.5 In equivocal, remote, or retrobulbar (i.e., not detectable by direct opthalmoscope) cases of optic neuritis, OCT can provide an objective and accurate measurement of retinal nerve fiber layer thickness. As discussed below, patients on Fingolimid (Gilenya) must be monitored for the development of macular edema, which can also be detected with this technology. Interestingly, axon loss in multiple sclerosis has been well demonstrated in the absence of clinical optic neuritis, thus prompting interest as a measure of overall disease activity and prognosis. OCT has not however been found to predict progression from isolated optic neuritis to clinically definite multiple sclerosis.
Advanced MRI techniques
Several MRI techniques under refinement will only increase the sensitivity and advance the timeline for detection of multiple sclerosis lesions. Magnetization transfer imaging (MTI) relies on the detection of decreased proton binding to macromolecules seen preferentially in early demyelinating lesions. This technique has generated interest as a more sensitive finding of both demyelination and remyelination than current gadolinium Enhancement. Diffusion Tensor Imaging (DTI) is able to detect abnormal directions of the diffusion of water molecules in MS lesions and has demonstrated clear abnormalities in white matter axon tracts which would have been considered normal with standard MRI techniques. Finally, Double Inversion Recovery (DIR) images, as the name implies, use two inversion pulses to differentiate with greater sensitivity cortical lesions (grey matter) from background white matter and cerebrospinal fluid.6
Selected Demyelinating Variants
Clinical Isolated Syndrome
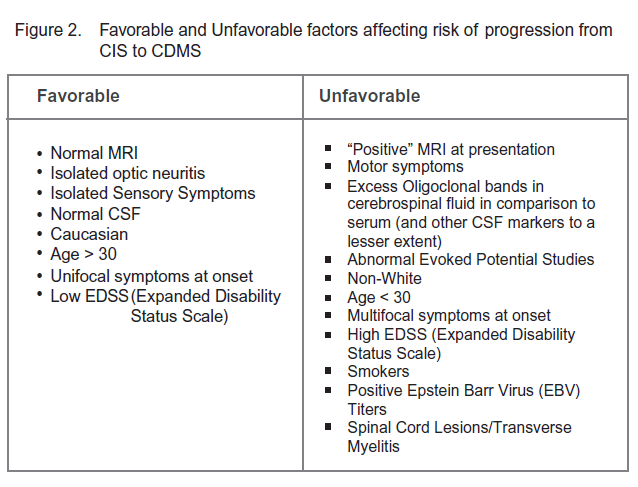
Clinically Isolated Syndrome (CIS) refers to a single clinical demyelinating event which then places the patient at higher risk for future development of clinically definite multiple sclerosis (CDMS). Underwriters should therefore be aware of the overall risk of progression to clinically definite multiple sclerosis as well as the prognostic factors which may significantly alter this risk. Estimates of the overall risk of developing multiple sclerosis vary widely depending upon cohorts and time frames studied, but is generally quoted at 50% within the next five years.8 Not surprisingly, the most important prognostic factor is often the MRI performed at the time of the clinical event. According to the National Multiple Sclerosis Society in the U.S., the risk of developing multiple sclerosis with a “positive” MRI for characteristic demyelinating lesions is between 60% and 80%.9 If MRI is normal at the time of the initial event, the risk drops to 20%.
Numerous studies8,10,11 have attempted to delineate further factors influencing the odds of progression to clinically definite multiple sclerosis. For ease these can be divided in to “favorable” and “unfavorable” factors (Figure 2).
 An important trend in the management of CIS is the shift toward treatment of a single demyelinating event with Disease Modifying Therapies (DMTs) previously reserved for CDMS. This reflects the findings of several key trials in CIS. The Controlled High Risk Avonex Multiple Sclerosis Study (CHAMPS) compared interferon beta 1a (Avonex) and placebo in a population of patients with CIS and at least two MRI lesions. The overall probability of developing CDMS was 35% in the interferon beta 1a group versus 50% in the placebo group at three years, a statistically significant difference (P=0.02).13,14The Early Treatment of Multiple Sclerosis (ETOMS) trial compared weekly interferon beta 1a (Rebif) with placebo in patients with CIS who met certain MRI criteria. The primary endpoint was time to CDMS: 569 days for interferon beta 1a versus 252 days for placebo (p = 0.034).13 In the BENEFIT Study (Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment), CIS patients were randomized to receive Interferon beta 1b (Betaseron) or placebo. At the end of two years, 28% of patients treated with DMT met criteria for CDMF versus 45% receiving placebo (p < 0.001)14 Finally, the Presenting With a Clinically Isolating Syndrome (PRECISE) assessed treatment with glatiramir acetate (Copaxone) versus placebo those patients with a combination of CIS and abnormal MRI, finding a 45% relative risk reduction in conversion rates to CDMS (p = 0.0005) by the end of the three-year study. Combining the results of these relatively short-term studies with similarly favorable extension studies has provided relative consensus favoring treatment of CIS (and abnormal MRI) with standard disease-modifying therapies. It can be anticipated that those evaluating critical illness claims will have to navigate an increasing population of claimants who do not yet meet formal criteria for CDMS but bear the same burden of injectable disease modifying treatments. It can be anticipated that those evaluating critical illness claims will have to navigate an increasing population of claimants who do not yet meet formal criteria for CDMS but bear the same burden of injectable disease modifying treatments.
An important trend in the management of CIS is the shift toward treatment of a single demyelinating event with Disease Modifying Therapies (DMTs) previously reserved for CDMS. This reflects the findings of several key trials in CIS. The Controlled High Risk Avonex Multiple Sclerosis Study (CHAMPS) compared interferon beta 1a (Avonex) and placebo in a population of patients with CIS and at least two MRI lesions. The overall probability of developing CDMS was 35% in the interferon beta 1a group versus 50% in the placebo group at three years, a statistically significant difference (P=0.02).13,14The Early Treatment of Multiple Sclerosis (ETOMS) trial compared weekly interferon beta 1a (Rebif) with placebo in patients with CIS who met certain MRI criteria. The primary endpoint was time to CDMS: 569 days for interferon beta 1a versus 252 days for placebo (p = 0.034).13 In the BENEFIT Study (Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment), CIS patients were randomized to receive Interferon beta 1b (Betaseron) or placebo. At the end of two years, 28% of patients treated with DMT met criteria for CDMF versus 45% receiving placebo (p < 0.001)14 Finally, the Presenting With a Clinically Isolating Syndrome (PRECISE) assessed treatment with glatiramir acetate (Copaxone) versus placebo those patients with a combination of CIS and abnormal MRI, finding a 45% relative risk reduction in conversion rates to CDMS (p = 0.0005) by the end of the three-year study. Combining the results of these relatively short-term studies with similarly favorable extension studies has provided relative consensus favoring treatment of CIS (and abnormal MRI) with standard disease-modifying therapies. It can be anticipated that those evaluating critical illness claims will have to navigate an increasing population of claimants who do not yet meet formal criteria for CDMS but bear the same burden of injectable disease modifying treatments. It can be anticipated that those evaluating critical illness claims will have to navigate an increasing population of claimants who do not yet meet formal criteria for CDMS but bear the same burden of injectable disease modifying treatments.
Neuromyelitis Optica (NMO)
Neuromyelitis Optica or “Devic’s Disease”, merits special attention as a demyelinating variant given recent advancements in the detection and understanding of its underlying pathology. NMO is increasingly thought of not as a single disorder but as a spectrum of disease unified by a combination of prominent spinal cord involvement and bilateral optic neuritis. The disorder differs from multiple sclerosis in several distinct ways. While individual attacks are often treated with intravenous methylprednisone (similar to MS), traditional DMTs have not been found to be helpful as primary maintenance therapy in NMO. In fact, several reports suggest that interferon beta agents may have an exacerbating effect on disease course. Common immunosuppressive agents utilized include Imuran, mycophenolate, and rituximab. In addition to corticosteroids, plasmapheresis is often attempted for aggressive or refractory clinical attacks. Further establishing NMO as a separate disorder was the discovery of specific associated antibodies to aquaporin-4 (AQP4) proteins located in the channels of the foot processes of astrocytes. With sensitivities and specificities estimated at 70% and 90% respectively,15 this commercially available serology has become a cornerstone of diagnosis. Finally, and perhaps most importantly, NMO is distinguished by a significantly worse prognosis than multiple sclerosis, although mortality estimates vary widely. In one of the largest series, Wingerchuk et al.16 reported a five-year survival rate of 68% for those with the more common relapsing form of the disease. In contrast, five-year survival rates for the monophasic form of the disease (maximal symptoms within one month of presentation) have been reported to be 90%.17
Newly Approved Therapies
September 2010 represented a revolution in the care of multiple sclerosis patients, with the approval of Fingolimid (Gilenya) by the U.S. FDA, the first oral disease-modifying therapy. Since that time, two other oral medications have been approved as maintenance therapy, Teriflunomide (Aubagio) and Dimethyl Fumurate (Tecfidera).
Fingolimid
Fingolimid acts as a sphingosine 1 phosphate receptor modulator. Although the exact mechanism is unclear, it is believed that binding of the compound to the receptor blocks the migration of lymphocytes from lymph nodes to the central nervous system, thus reducing the autoimmune response of multiple sclerosis. Two pivotal phase III trials prompted approval of fingolimid in multiple sclerosis. In the FREEDOMS study (FTY720 Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis), fingolimid was compared with placebo. Fingolimid met the primary endpoint, a reduction in the annual relapse rate (0.16 for fingolimid versus 0.40 for placebo, p<0.01), correlating to a 60% relative reduction. Secondary endpoints of time to disability progression and MRI lesion burden (the number of gadolinium enhancing lesions) were also met.18 Fingolimid was also compared with interferon beta 1a in the Trial Assessing Injectable Interferon Versus FTY720 Oral in Relapsing-Remitting Multiple Sclerosis (TRANSFORMS) study. Annualized relapse rate again was the primary endpoint and was significantly lower in the 0.5 mg fingolimid group (0.16) versus interferon beta-1a (0.33, p<0.01). Statistically significant differences in MRI lesions were also found but there was no significant difference in disability progression, likely secondary to a low rate overall of disability progression in both groups.19
Despite this promising data, several important side effects have emerged from analysis of fingolimid studies. Though rare, cardiac adverse events have been noted, particularly with the first dose. In the two previously mentioned studies, the rates of first-degree AV block and mobitz type I AV blocks were estimated to be 4.7% and 0.2 respectively.20 After report of a first-dose cardiac death on the medication, it is recommended that patients be monitored in the office for six hours after initially receiving the medication. Patients with cardiac comorbidities are often monitored overnight in a hospital setting. An additional unique concern with the medication is macular edema. Again, although rare (0.2 to 1.1% depending on fingolimid dose),20 it is recommended that patients have ophthalmology exams (often utilizing the previously mentioned OCT technology) prior to the medication and for several months thereafter to detect this condition. Finally, there have been case reports of fatal herpes encephalitis, Progressive Multifocal Leukoencephalopathy (PML), and Varicella Zoster (VZV) in association with the medication though causality is controversial.
Teriflunomide
Teriflunomide was FDA approved for the treatment of multiple sclerosis in September of 2012. The oral medication’s mechanism of action in multiple sclerosis is unclear, although it is known to block pyrimidine synthesis. In the first phase III study, Teriflunomide Mutiple Sclerosis Oral Trial (TEMSO), teriflunomide reduced the annual relapse rate in comparison to placebo (0.37 teriflunomide versus 0.54 placebo, p < 0.001). At higher doses (14 mg) further improvements were seen in disease progression rates and MRI metrics.21 In a subsequent comparison with interferon beta 1a, the Teriflunomide and Rebif (TENERE) Trial, there was no statistically significant difference between teriflunomide and interferon beta 1a in the somewhat unusual primary endpoint of treatment failure (defined as either relapse or discontinuation). The medication does come with two ‘black box’ warnings: 1) Increased ALT levels are described in roughly 14% of cases22 with case reports of liver failure. This prompted recommendations to avoid the medication in those with pre-existing liver disease and follow levels for six months for all others. 2) The medication is contraindicated in woman of childbearing age, given reports of teratogenicity in animal models.
Dimethyl fumarate
Dimethyl fumarate is an oral compound also known by its previous name, BG-12. A chemically related compound has been used for several years to treat psoriasis. Once again, the exact mechanism of the medication remains unclear, but given its role in the activation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway, an anti-oxidative and neuroprotective pathway has been proposed. Dimethyl Fumarate was FDA approved for the treatment of multiple sclerosis base on two randomized controlled phase III studies, the DEFINE (Determination of the Efficacy and Safety of Oral Fumarate in RRMS) and CONFIRM (Comparator and an Oral Fumarate in RRMS) studies. In the DEFINE study, dimethyl fumarate was compared with placebo; the primary endpoint was the percentage of patient who experienced a relapse at two years. This proportion was significantly lower with dimethyl fumarate than placebo (27% versus 46%, p<0.01) corresponding annualized relapse rates of 0.17 and 0.36 respectively.23 In the CONFIRM study, dimethyl fumarate and a common injectable, glatiramer acetate (Copaxone), were compared with placebo. This study confirmed superiority over placebo (annual relapse rate 0.20 dimethyl fumarate versus 0.40 placebo, p< 0.001), which was the primary design goal. Subsequent analysis, however, did not show a statistically significant difference between dimethyl fumarate and glatiramer acetate.24 Case reports of PML have been described in association with dimethyl fumarate and other fumarate compounds both in patients with multiple sclerosis and psoriasis. Lymphopenia is common with the medication: the pattern is a 30% drop in the first year followed by stabilization; therefore, it is recommended that complete blood cell counts be followed while on therapy. Rates of serious infection however have not been found to be more common on the medication.25
Alemtuzumab
Alemtuzumab was approved in Europe in September 2013 as a first-line agent in the treatment of multiple sclerosis. A few months later, the FDA rejected approval in the medication in a surprising and controversial decision (the medication had previously been granted fast-track designation). Appeal of the rejection is ongoing. The medication is a monoclonal antibody directed against the CD52 antigen which results in significant and prolonged lymphocyte depletion. One potentially very appealing aspect of the medication is its dosing schedule. In trials it was administered as a single five-day intravenous infusion followed by a three-day infusion one year later. In the CARE MS I trial alemtuzumab was compared with interferon beta 1a. Primary endpoint points were relatively aggressively chosen: an improved relapse rate in comparison with interferon beta 1a (generally considered to be ‘strongest’ injectable disease modifying therapy) and six-month disability scores. The medication was able to meet the first primary endpoint (annualized relapse rates 0.18 for alemtuzumab and 0.39 for interferon beta 1a, p<0.0001) but not the second.26,27 The subsequent CARE MS II trial showed similar findings in a cohort of patients who had failed previous disease modifying therapies.28 The rejection by the FDA was based upon concerns with study design but also safety concerns, in particular a high rate of autoimmune disorders. In pooled analysis autoimmund thyroid complications were seen in 18% of patients, 21 cases of ITP and four cases of glomerulonephritis.29
Conclusion
The landscape of multiple sclerosis is changing. Patients are being diagnosed and treated earlier, with a much wider range of options. Long-term data on these agents is lacking both in terms of mortality and adverse events. Prospects for mortality improvements however are auspicious given initial data on relapse rates and MRI lesion burdens with newly approved agents. Compliance rates are expected to significantly improve with (for many) the replacement of injectables with oral therapies. Moreover, the increasing range of options will likely lower the threshold for changing the regimen of a patient who is relapsing on current therapies. Combinations of therapies may provide additional benefit but this is being navigated cautiously given that most cases of Progressive Multifocal Leukoencephalopathy (PML) have occurred in patients on multiple immunomodulatory agents. Accurate diagnosis, a highly tailored approach, and disease remission remain paramount goals.