Sudden cardiac death (SCD) is the most common cause of death in Western countries.11
The incidence of sudden death in the general population in Europe is 1.34:100,000 for the ages 7 - 64, with 5 - 8% of those showing no evidence of any structural cardiac abnormality or evidence of any coronary disease at autopsy.1,2
Unexpected sudden cardiac death may occur at any age and results from the development of a malignant aberrant rhythm – ventricular tachycardia (VT) or ventricular fibrillation (VF) – occurring either on a background of structural heart disease, including coronary artery disease, or in a heart that at autopsy is deemed to be structurally normal.
Sudden cardiac death may be aborted if an arrhythmia is very brief and self-terminating but death may otherwise be inevitable in the absence of emergency resuscitation. Sudden cardiac death underlies about 20% of total mortality and 50% of cardiovascular mortality in Western countries. It is the most frequent mode of death during exercise, with 75% of deaths during exercise being cardiac-related.
Heart disease is the most common cause of sudden death in young athletes where physiological demands, coupled with an occult cardiac condition, poses an inherent mortality risk which is greater than in age-matched sedentary individuals.
Coronary artery disease and non-coronary structural heart disease are the most common causes of sudden cardiac arrest. Those structural conditions responsible for sudden arrhythmic death include myocardial infarction and ischemia, valvular heart disease, congenital heart disease and cardiomyopathy.
Ischemic heart disease is the most common structural heart condition responsible for sudden cardiac death in older age groups, while in young people aged <35 heritable hypertrophic cardiomyopathy and other congenital / heritable structural cardiac abnormalities predominate.
Cardiomyopathy may be congenital / heritable or acquired and includes hypertrophic cardiomyopathy, ischemic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and dilated cardiomyopathy.
Inborn errors thus have the most impact on SCD in the young while acquired factors dominate risk in older subjects.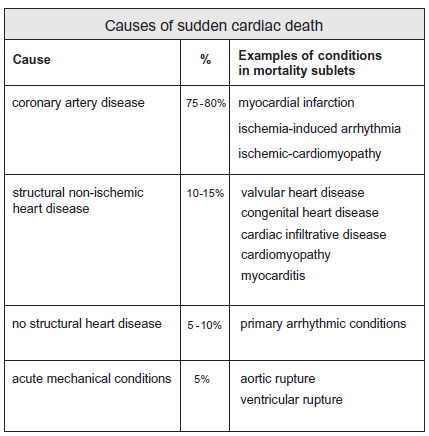
This review focuses on sudden cardiac death in those with structurally normal hearts where sudden death occurs in the absence of any overt heart disease at a comprehensive post mortem.
No structural abnormality is detectable in perhaps 5 - 10% of sudden cardiac deaths and these deaths are deemed to be arrhythmogenic. Sudden arrhythmic death is believed to be responsible for approximately 14% of sudden cardiac deaths in young people in the U.K.
These primary lethal rhythm disturbances most commonly result from the presence of an inherited congenital arrhythmogenic disease. These conditions usually have a diagnostic signature on the surface electrocardiogram.
As a group, these conditions are deemed channelopathies, given that they arise as a consequence of abnormalities in the ion channels (sodium, potassium or calcium transport channels) that are responsible for electrical conduction in the heart.
In the past couple of decades a growing number of distinct and potentially lethal heritable channelopathies have been described and include:
- Long QT syndrome (LQTS)
- Short QT syndrome (SQTS)
- Brugada syndrome (BrS)
- Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
The genetics of these syndromes are complex. The currently known genetic abnormalities associated with the channelopathies are not detected in all those who have been resuscitated from sudden death or in the deceased who have been submitted to a genetic autopsy.
The Brugada syndrome is probably the most well-known channelopathy, having been described as a clinical entity by the brothers Drs. Pedro, Josep and Ramon Brugada in 1992 as a result of the classical ECG abnormality (previously considered a normal variant) being linked to recurrent syncope in a three-year-old Polish boy whose sister had died suddenly at two years of age. It has attracted a great deal of interest as a risk factor for sudden death because of its high incidence in some parts of the world.
While the Brugada syndrome is a congenital abnormality, the symptoms, including sudden death during sleep, can develop for the first time at any age from infancy to old age, with the mean age of onset of symptomatic electrical events being age 40±22.5
Symptomatic events with syncope or resuscitated cardiac death relate to the abrupt onset of polymorphic ventricular tachycardia or ventricular fibrillation without any prodrome. Brugada is also associated with high rates of paroxysmal atrial fibrillation and other supraventricular arrhythmias with prevalence rates of about 20%.3,5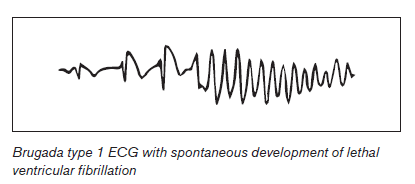
The Brugada syndrome is believed to be responsible for 4 - 12% of all unexplained sudden death and 20% of sudden death in those with structurally normal hearts at autopsy in some series.9,4
The Brugada syndrome, defined as symptoms in an individual with a Brugada ECG pattern, has a prevalence of 1-5:10,000 worldwide and a prevalence of ≥5:10,000 in Southeast Asia.5 In Southeast Asia, the Brugada syndrome is the leading cause of death in males under 40 years of age. The Asian prevalence of a Brugada ECG pattern may be as high as 2% in some areas compared to a European prevalence of 0% - 0.8% and a U.S. prevalence of 0.02%. Prevalence rates are highest in Thailand and the Philippines.1,5 Brugada is a single gene disease inherited in an autosomal dominant fashion and 20 - 50% will have a family history of sudden death. Up to 60% of Brugada may be sporadic, with no evidence of the disease in a parent or in other relatives, although not all with Brugada will have expressed the phenotype. Some people with a Brugada ECG will remain asymptomatic throughout their lifetimes.4
The expression of the arrhythmic phenotype in those with a Brugada ECG may be modulated by the presence of environmental factors, which include fever and certain medications. Genetic factors may also play a role in the promotion of arrhythmias in those with a Brugada ECG pattern.
Although the inheritance is autosomal dominant there is a marked male predominance of the arrhythmic phenotype with a male to female ratio of approximately 8:1. It is not clear why the phenotype is more penetrant in males with a Brugada ECG than in females.
The diagnostic criteria for the Brugada syndrome require two components:
1. Detection of the characteristic ECG pattern plus
2. Clinical characteristics
Three Brugada patterns on the ECG are described (types I, II and III) but a Brugada syndrome is only definitively diagnosed when a type I coved shaped ST segment pattern is identified in the context of one of the following associated characteristics.10
- Syncope or resuscitated sudden cardiac death
- Nocturnal agonal respiration
- Documented VT or VF
- A family history of sudden cardiac death aged <45
- Type I ECGs in family members
- Inducible VT at electrophysiological study
The type I pattern may be dynamic, temporally variable or concealed making the true prevalence of Brugada syndrome difficult to estimate. The ECG pattern can vary spontaneously and may even disappear periodically.12 The type I diagnostic pattern may occur spontaneously or be unmasked by a drug challenge in the laboratory. A type I pattern may also be unmasked by fever, metabolic conditions, oral medications and anesthetic drugs. ECG type II (a saddleback ST segment configuration with >= 1mm ST elevation) and type III ( a saddleback ST segment configuration with <1mm ST elevation) are the most prevalent Brugada ECG patterns in all populations and should not be considered diagnostic of the Brugada syndrome. The type I pattern is far less frequent.
A drug challenge in the laboratory is usually mandated in any person with a type II or type III ECG in the context of symptoms or other high risk features, with the intent being to determine whether or not a change in the ECG morphology to a type I can be induced.
A diagnosis of Brugada syndrome can be made when a type II or type III ECG pattern is present in baseline conditions and where there is a conversion to the diagnostic type I ECG after drug provocation provided that one of the non-ECG components of the syndrome is present.3
First-line therapy for people with a Brugada syndrome involves the deployment of an implantable defibrillator (ICD) although there are drugs that have the capacity to suppress ventricular arrhythmias. Several studies have demonstrated the vast superiority of device therapy over antiarrhythmic medications in this population so that drug therapy is not considered appropriate treatment.
Defibrillator deployment is mandated in individuals with aborted sudden death or syncope of cardiac origin in the context of a spontaneous type I pattern.
Strong indications for device deployment also include:
1. Aborted sudden death or syncope in an individual with an inducible type I pattern
2. Inducible VT / VF in an asymptomatic individual with a spontaneous type I pattern
From an insurance perspective, the mortality expectations of Brugada syndrome may preclude an offer, even in the context of device therapy. Despite the protective effects of defibrillators, it is important to realize that ICDs have significant complication rates particularly when balanced against the low annual rates of arrhythmic events even when ICDs have been deployed in persons with symptoms.
The deployment of ICDs for type I ECG patterns in populations with resuscitated sudden death, syncope or positive electrophysiological studies has been associated with device-related complication rates as high as 8.9% per annum. This with associated annual arrhythmic event rates of only 2.6% per annum (1.5% in asymptomatic people).13
Device complications include inappropriate shocks (with inappropriate shocks being 2.5 times more frequent than appropriate ones), venous thrombosis and pulmonary embolism, pericardial effusions, lead failure, device re-implantation and severe psychological difficulties.13
There has been a large focus on the issue of risk stratification in patients at risk of sudden death, although those with aborted sudden cardiac arrest are clearly at the highest end of the risk spectrum and require the protection of a defibrillator.
Stratification in the context of a Brugada ECG in an asymptomatic individual with no adverse family history is very difficult.
There is considerable disparity in the data regarding the incidence of life threatening-events in individuals with Brugada ECG criteria. The initial studies by Brugada in 1998 reported annual rates of first events of 10% but this study almost certainly included only higher risk individuals. The same authors in 2002 reported annual first event rates of 3.5%.
In 2005, Brugada reported first VF event rates of 1.7% annually. Priori et al. in 2002 reported cumulative cardiac arrest rates by age 40 of 14% in asymptomatic individuals, corresponding to an annual incidence of 0.35%. That same group in 2005 reported annual rates of 1%.
Eckardt 2005 reported a first event in 0.8% of individuals with type I Brugada ECGs followed over a 40±50 month period corresponding to an annual event rate of 0.24%, much lower than in other studies.8
The reasons behind the apparent progressive decline in annual event rates over time is not clear but probably results from the inclusion of lower risk phenotypes in some studies and the inclusion of type II and type III in the Priori data.5,6,7
In the most recent review of the international Brugada registry the lifetime risk of sudden death or ventricular fibrillation is reported to be 25% (mean age 42±15), with most individuals remaining asymptomatic. Reports from other series describe lifetime rates of syncope or sudden death in 17-42% of diagnosed individuals.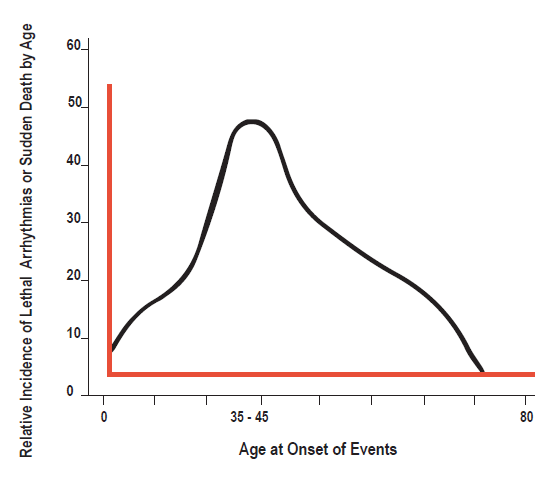
Previous syncope has been reported in up to 23% of those with cardiac arrest.16
Event rates are higher in Asian populations with Brugada ECGs than in European populations suggesting differing genetic predispositions.
Psychotropics, anesthetics, antihistamines, cocaine and other drugs are well-documented precipitants of the type I pattern and may induce symptoms. Medications thus increase the risk of penetration of the syndrome in an otherwise asymptomatic person regardless of the ECG morphology.2
The lack of symptoms at older ages in an applicant does not protect against sudden death or arrhythmogenic syncope.
Asymptomatic individuals with no symptoms and a negative family history are not necessarily at low risk.11 Those with a spontaneous type I pattern are at a higher risk than those where a type I morphology has only been exposed by a drug challenge.2,14 Individuals with a type I ECG and symptoms have a RR = 6 of death compared to those with a type I ECG and no symptoms. There is no good evidence that a lack of a family history is sufficiently protective: asymptomatic individuals with a characteristic ECG and no family history are not necessarily at low risk. The serendipitous finding of a spontaneous type I Brugada ECG in an applicant might lead to a decline, although the literature suggests a high negative predictive value (93%) for events in asymptomatic people if arrhythmias are not inducible at an electrophysiological study.1,15 The annual event rates in asymptomatic people with a type I pattern reported by Obeyesekere in 2011 was approximately 0.5% compared with annual event rates of 7.7% in those with aborted SCD.2
Brugada reported recurrence rates of about 20% in aborted SCD in type I ECG.9 Approximately one third of asymptomatic type I individuals will have inducible VT /VF at an electrophysiological study but there is there is conflict in the data as to whether inducability predicts the risk of spontaneous VT/VF. From an insurance perspective a negative VT/VF provocation study in a type I morphology does not mitigate risk despite the negative predictive value.
The chance finding of a type II or type III pattern in an asymptomatic applicant may allow terms if there have been no symptoms and particularly if there is no adverse family history. In completely asymptomatic individuals with only drug inducible type I patterns the event rates are low.1 Clinical practice guidelines do not mandate drug provocation testing in non-type I patterns in people with no symptoms as this does not appear to add value to risk stratification. There is nonetheless a view that this approach is useful in terms of providing advice on lifestyle, fever management, medication use and the management of syncope.
The Brugada ECG signature and the ECG signatures of other occult syndromes that have the potential to induce malignant arrhythmias have important mortality implications. This supports the utility of the ECG at underwriting, particularly in Southeast Asian and other high-risk populations.
