Executive Summary
- The pathogenesis of COVID-19 infection drives treatment guidelines.
- For most non-hospitalized adults at risk for progression to more severe COVID-19, preferred treatment options are antivirals Paxlovid and remdesivir, with the alternative treatment of Bebtelovimab (a monoclonal antibody) and the antiviral molnupiravir in cases where neither of the preferred therapies are available, feasible to use or clinically appropriate.
- One important concept to understand is the distinction between FDA approval and the designation of Emergency Use Authorization (EUA) since most of the available SARS-CoV-2 treatments are available under an EUA.
- During times of public health emergency, including infectious diseases such as the SARS-CoV-2 pandemic, the EUA may be used to allow for the use of unapproved medical products or unapproved uses of approved medical products without full FDA approval to accelerate the timeline.
- This article provides a description of the FDA approval process, the use of the EUA, a timeline of approval for SAR-CoV-2 treatments, and a descriptive summary table with available treatments for underwriting reference.
The Pathophysiology of COVID-19
The pathogenesis of COVID-19 infection drives treatment guidelines: early in the clinical course, the replication of SARS-CoV-2 in the upper respiratory tract (nasopharynx, oropharynx, trachea) leads to infection and inflammation, triggering an immune response. If the virus is not cleared by innate or adaptive responses, it can spread to the lower respiratory tract by inhalation of the virus particles from the upper respiratory tract or by gradual dissemination along the tracheobronchial tree. Alternatively, the initial site of infection can be the lower respiratory tract. This can ultimately lead to infection of the alveoli, causing inflammation and limiting gas exchange,[1] resulting in pneumonia and hypoxia.
Later in the clinical course, the disease appears to be driven by a dysregulated immune/inflammatory response to SARS-CoV-2 that leads to tissue damage.[2] Shortness of breath and hypoxia can occur, leading to acute respiratory failure and ARDS. Other hyper-inflammatory responses can lead to extrapulmonary disease, such as acute kidney failure, myocarditis, coagulopathy and acute liver injury. Therefore, therapies that directly target SARS-CoV-2 would have the greatest effect early in the course of the dis- ease, while immunosuppressive/anti-inflammatory therapies are likely to be more beneficial in the later stages of COVID-19.[2] Examples of immunosuppressive medications are steroids such as dexamethasone and immunomodulatory drugs such as baricitinib and tocilizumab.
Clinical Treatment Guidelines
Treatment guidelines vary depending on severity of disease, age and comorbidities of the affected individual, as well as outpatient vs. hospitalization status. Disease severity is defined as mild, moderate and severe, with mild disease manifesting as cough, malaise and upper respiratory symptoms; moderate disease with dyspnea; and severe disease with hypoxemia requiring oxygen or ventilatory support. Those with severe disease require hospitalization and/or intensive care unit (ICU) care.
Per the most current recommendations of the National Institutes of Health COVID-19 Treatment Guidelines Panel,[2] non-hospitalized adults who are at risk of progressing to severe COVID-19 are those who are older and/or with an underlying medical condition such as cancer, chronic lung diseases, cardiovascular disease, diabetes, chronic liver disease and kidney disease.[3] For these individuals, preferred treatment options are antivirals Paxlovid and remdesivir, with the alternative treatment of Bebtelovimab (a mono- clonal antibody) and the antiviral molnupiravir in cases where neither of the preferred therapies are available, feasible to use or clinically appropriate.
Treatment for hospitalized adults is stratified based on disease severity:[2]
- For those who do not require supplemental oxygen, if at high risk of disease progression, remdesivir may be appropriate.
- For those who require supplemental oxygen, remdesivir or a combination of remdesivir and dexamethasone is recommended, with the addition of an immunomodulatory drug for those with rapidly increasing oxygen needs and systemic inflammation.
- For those who require oxygen through a high-flow device or non-invasive ventilation, dexamethasone or a combination of dexamethasone plus remdesivir is recommended, with the addition of an immunomodulatory drug for those with rapidly increasing oxygen needs and systemic inflammation.
- For those who require mechanical ventilation or extracorporeal membrane oxygenation (ECMO), dexamethasone is recommended, with an addition of immunomodulatory drug if within 24 hours of admission to the ICU.
Management of COVID-19 in children is considered on a case-by-case basis, following a similar practice of directed antiviral treatment, adjusted for age, for those who are managed as outpatients, and immunosuppressive medications for hospitalized individuals. Multisystem inflammatory syndrome in children (MISC-C), a rare and severe manifestation of COVID-19 resulting in shock, cardiopulmonary, and neurocognitive signs and symptoms, requires a separate consideration for treatment and management.
Therefore, these guidelines reflect the pathophysiology of COVID-19 infection targeting the virus, in which early treatment and treatment of mild to moderate infection are composed of antiviral medications and monoclonal antibodies, while treatment of severe disease and late stages of the infection include medications that suppress the immune system to control a hyperactive and dysregulated immune response.
SARS-CoV-2 Treatment
FDA Approval vs. EUA
One important concept to understand is the distinction between FDA approval and the designation of EUA, since most of the available SARS-CoV-2 treatments are available under an EUA. The traditional pathway to FDA approval can take more than a de- cade,[4] which is a significant obstacle during a public health emergency. The process of moving a drug from development to FDA approval begins with preclinical testing in an animal model. Most drugs never make it out of this stage of testing. When a drug shows promise in the preclinical stage, the sponsor institution submits an Investigational New Drug Application (IND) to the FDA.[5] In this application, the sponsor must show results of the preclinical testing and the plan for human testing. If both FDA and the Institutional Review Boards (IRBs) from all of the involved institutions (drug companies, hospitals, universities) determine that it is safe to move forward, then Phase 1 clinical trials can begin.
In Phase 1, the goal is safety. Phase 1 studies are conducted to determine the drug’s side effects and metabolism. The study population is usually healthy adults (n<100 people). Assuming the drug is deter- mined to be safe, then it moves to Phase 2 studies, with the goal of determining if the drug is effective. The study design in Phase 2 might compare the effect of the new drug in patients with the target disease to those taking an existing treatment or to those taking a placebo. Both effectiveness and safety are being evaluated and the number of patients is still small (n<300 people). If the Phase 2 data shows that the drug is safe and effective, then it can move on to Phase 3.5 In Phase 3, effectiveness needs to be shown in larger populations of people, in different populations, at various doses and in combination with other drugs. Safety and effectiveness are still the primary goal. This phase may involve several thousand people. Assuming everything goes well in Phase 3, the sponsor can submit a New Drug Application (NDA) to the FDA. This submission is a formal request for approval of the drug in the US and contains all data about the drug from initial development through clinical trials (Phases 1-3). The FDA has 60 days to decide whether to review the NDA. Once the NDA is filed, meaning the FDA review will move forward, a team of experts is assembled. They review the submitted study data in the NDA and each reviewer prepares an evaluation with recommendations. The FDA may also convene an advisory committee to provide independent opinions about the drug. The drug must show clinical benefit to receive traditional approval. Then the drug will either be approved, or the sponsor will receive a response letter. Some drugs are given accelerated approval if they target a life-threatening disease or condition with no other available treatments. If the drug is approved, Phase 4, or post-marketing surveil- lance data, is collected to look for additional safety issues that may emerge as a much larger population takes the new drug.[4,5,6]
During times of public health emergency, including infectious diseases such as the SARS-CoV-2 pandemic, the EUA may be used to allow for the use of unapproved medical products or unapproved uses of approved medical products without full FDA approval, to accelerate the timeline. The Secretary of HHS must declare that an EUA is appropriate, which occurred on February 4, 2020, for SARS-CoV-2.[7] The benefits of the drug or treatment being considered for the EUA still must outweigh any known risks, and there must not be any alternative available. EUAs end when the public health emergency ends, but they can also be terminated at any time (JHU ctr health security). Most of the available treatments for SARS-CoV-2 are currently being used under an EUA. This has allowed accelerated movement of treatments to the public that show evidence of benefit but have not met the full criteria for FDA approval. Some of these treatments may go on to gather enough data to gain full FDA approval, as happened with remdesivir, which was originally authorized for use under the EUA and gained full FDA approval in October 2020 (see Figure 1 below). Others may be found to be less effective than early data showed, and the EUA will be terminated, as was the case for several drugs that were not effective against the Omicron variant (REGEN-COV, sotrovimab) or were found to be ineffective against Omicron and had additional safety concerns (bamlanivimab).[2]
Timeline of Treatment Availability for SARS-CoV-2
Figure 1 below shows a timeline of treatment approvals for SARS-CoV-2. The first treatments specific to SARS-CoV-2 became available in May 2020 under an EUA. Remdesvir, an antiviral with US brand name Veklury (Gilead Sciences, Inc.), was the first drug to receive full FDA approval for treatment of SARS- CoV-2. Previously, under an EUA from May 1, 2020, Remdesvir had been authorized for use in a larger population of patients.[8]
Figure 1. COVID-19 Treatment Approvals
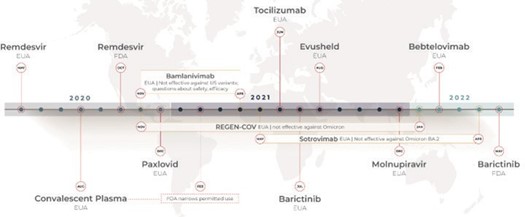
Table 1 contains a summary of the available treatments.[9] Further discussion of the preferred treatments continues below.
Table 1. Available Treatments - Summary Table
Remdesivir
Remdesivir, the antiviral with brand name Veklury and study drug name GS-5734, was evaluated in three clinical trials. In a Phase 3, open-label, randomized trial (Study 5773), hospitalized patients (n=397) with confirmed COVID-19, oxygen saturation ≤94% breathing ambient air (not requiring mechanical ventilation) and pneumonia were randomly assigned to receive remdesivir treatment for 5 or 10 days. The goal of the trial was to determine if a 5-day course of treatment would be as safe and effective as a 10-day course of treatment in patients with severe COVID-19. Patients receiving 10 days of treatment were not significantly different in clinical status compared to those receiving 5 days of treatment at the conclusion of the trial (p=0.14).[10,11]
In another Phase 3, randomized, open-label trial, hospitalized patients with moderate COVID-19 were randomized to a 5-day or 10-day course of treatment with remdesivir. The goal of this trial was to determine if 5 days of treatment was as effective as 10 days in those with moderate COVID-19, defined as hospitalized individuals with a confirmed COVID-19 diagnosis, pneumonia and oxygen saturation >94% on room air. The 596 patients were randomized to receive standard of care or the 5 or 10-day treatment with remdesivir. At the end of the trial, those receiving 5 days of treatment showed improvement compared to those in the standard of care group (p-0.02), while those in the 10-day treatment group did not show a statistically significant difference in clinical status (p=0.18).[10,12]
Finally, the ACTT-1 study was a randomized, double- blind, placebo-controlled trial with 1,062 patients. Adults hospitalized with COVID-19 showing evidence of lower respiratory tract infection were randomly assigned to receive either remdesivir (n=541) or placebo (n=521). Those in the remdesivir group had a median recovery time of 10 days compared to those in the placebo group, with a median recovery time of 15 days (p<0.001).[10,13]
Paxlovid
For Paxlovid, also known as nirmatrelvir plus ritonavir, the main data come from the EPIC-HR trial. This Phase 2/3, randomized, double-blind, placebo- controlled study was designed to evaluate safety and efficacy of nirmatrelvir plus ritonavir in non-hospitalized adults with mild to moderate COVID-19 who were at risk of progression to more severe disease. Patients (n=2,246) were randomized to receive either nirmatrelvir plus ritonavir or placebo within 3 days of symptom onset over a 5-day period. The outcomes measured were hospitalization and all-cause mortality. At the conclusion of the trial, day 28, the incidence of hospitalization was lower in the treatment group compared to the placebo group (p<0.001), and there were zero deaths in the treatment group compared to 7% mortality in the placebo group. Paxlovid is available under an EUA.[14,15,16]
Conclusion
Clinical treatment guidelines of COVID-19 reflect the pathophysiology of the disease process. Research and development for new therapies for SARS-CoV-19 are ongoing as are clinical trials of existing medications, many of which have shown promise. As described, the process of moving a potential treatment from the laboratory to the clinic is a long one, even under accelerated approval and EUA. As additional data is gathered on the existing treatments, some may move to full FDA approval while others may prove not to be as effective as initially hope